|
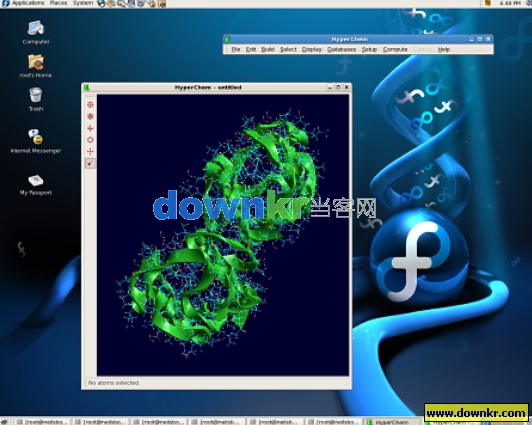
Hyperchem是一款以质量高,灵活易操作而闻名的分子模拟软件。通过3D对量子化学计算,分子力学及动力学进行模拟。Hyperchem为您提供比其它Windows软件更多的模拟工具: 图形界面,半经验计算方法(AM1,PM3等),UHF,RHF和CI,7.0版新增加的密度泛函。可进行单点能,几何优化,分子轨道分析,蒙特卡罗和分子力学计算,预测可见-紫外光谱。
功能
1. 结构输入和对分子操作。
2. 显示分子。
3. 化学计算。用量子化学或经典势能曲面方法,进行单点丶几何优化和过渡态寻找计算。可以进行的计算类型有:单点能,几何优化,计算振动频率得到简正模式,过渡态寻找,分子动力学模拟,Langevin动力学模拟,Metropolis Monte Carlo模拟。支持的计算方法有:从头计算,半经验方法,分子力学,混合计算等。
4. 可以用来研究的分子特性有:同位素的相对稳定性,生成热,活化能,原子电荷,HOMO-LUMO能隙,电离势,电子亲和力,偶极矩,电子能级,MP2电子相关能,CI激发态能量,过渡态结构和能量,非键相互作用能,UV-VIS吸收谱,IR吸收谱,同位素对振动的影响,对结构特性的碰撞影响,团簇的稳定性。
5. 支持用户定制的外部程序。
6. 其它模块:RAYTRACE模块,RMS Fit,SEQUENCE编辑器,晶体构造器糖类构造器,构像搜寻,QSAR特性,脚本编辑器。
7. 新的力场方法:Amber 2,Amber 3,用于糖类的Amber,Amber 94,Amber 96。
8. ESR谱。
9. 电极化率。
10. 二维和三维势能图。
11. 蛋白质设计。
12. 电场。
13. 梯度的图形显示。
14. 新增功能:密度泛函理论(DFT)计算,NMR模拟,数据库,Charmm蛋白质模拟,半经验方法TNDO,磁场中分子计算,激发态几何优化,MP2相关结构优化,新的芳香环图,交互式参数控制,增强的聚合物构造功能,新增基组。
|